
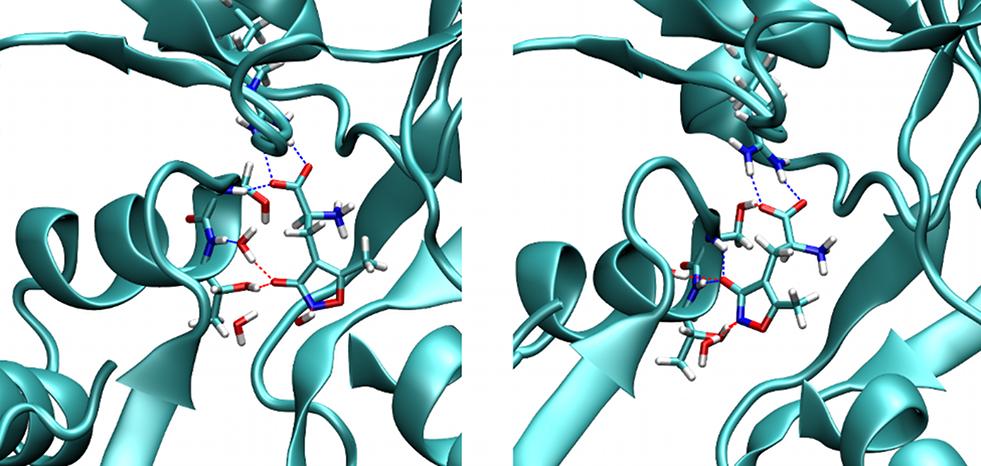
The aim of molecular modeling glutamate receptors is to support experimental activities in the design of new photo-switchable ligands for these ionotropic receptors as well as to provide an understanding in atomic detail of how such ligands interact with the receptors. The main questions involves computing ligand binding free energies and pathways, the maximal absorption wavelength of a ligand and the mechanism of photo-isomerization. Our work encompasses three steps:
(i) Predicting binding modes for known ligands. We could show that standard ligand docking algorithms did not lead to reliable results for the open clam-shell receptor, due to the complexity of the accompanying protein conformational change. Therefore we established a more complex procedure of rigorous free energy calculations using the known AMPA ligand.
(ii) Using this methodology, we investigate the binding behavior of new ligands synthesized in the Trauner group. Our approach allows us to distinguish different contributions to the binding strength stemming from protein conformational changes and from differences in ligand structure.
(iii) Finally, we used our validated binding mode prediction to understand the photo-switching mechanism of the ligand, i.e. to understand how light induced ligand structure changes induce the channel opening.