
Machine-Learning Potentials for QM/MM
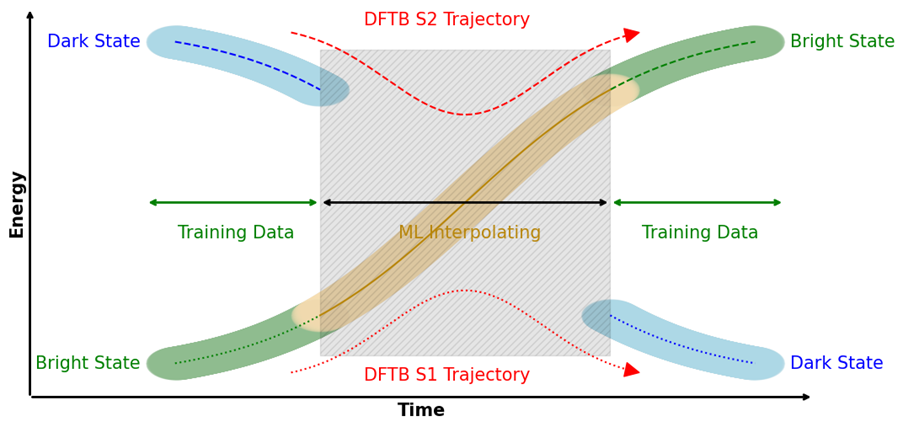
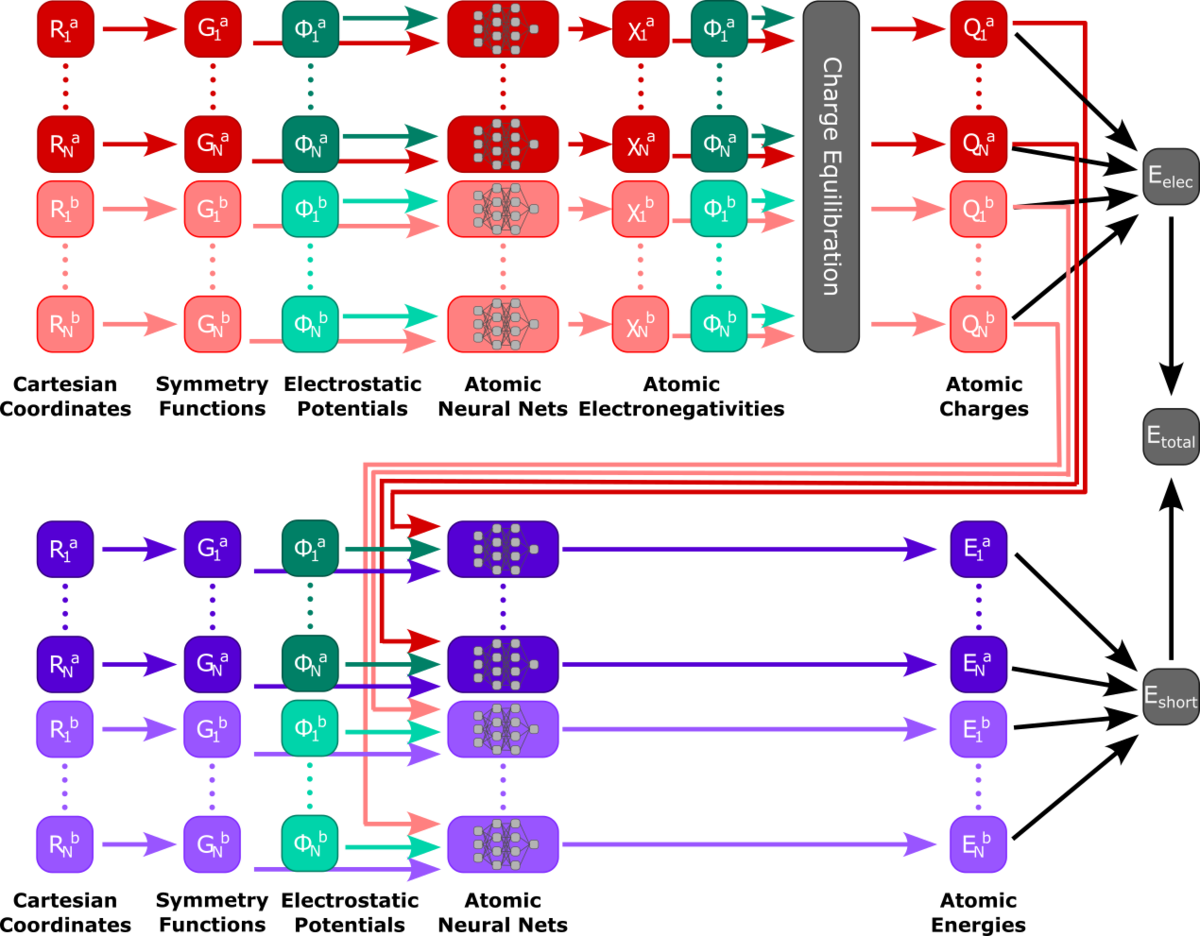
Machine learning potentials (MLPs) offer new possibilities for molecular dynamics simulations and property calculations. MLP-driven molecular dynamics simulations are an attractive alternative to both ab initio methods, because of their computational performance, and semi-empirical methods, because of their adaptability to custom data. Automatic differentiation in powerful libraries enables the rapid parameterization of system-specific and transferable models alike, giving rise to various recent developments of model architecture like HDNNPs, Schnet, MACE, and many others. Our own research interests focus especially on the combination of molecular environments and MLPs in a QM/MM fashion, excited state MLPs, fluorescence properties, and efficient sampling methods for the training of MLPs.
Light-Harvesting In Photosynthetic Proteins

Life as we know it depends on photosynthesis—a process that converts sunlight into energy-rich molecules. While we understand what happens during photosynthesis, the how—the intricate details of its light-driven mechanisms—remains a fascinating puzzle.
At its core, photosynthesis involves:
1. Capturing sunlight via pigments in light-harvesting complexes (LHCs)
2. Transferring this energy to reaction centers
3. Initiating charge separation at the reaction center
4. Generating a proton gradient using the separated charge
5. Converting the proton gradient to chemical energy (ATP)
Proton Coupled Electron Transfer in Biomolecules
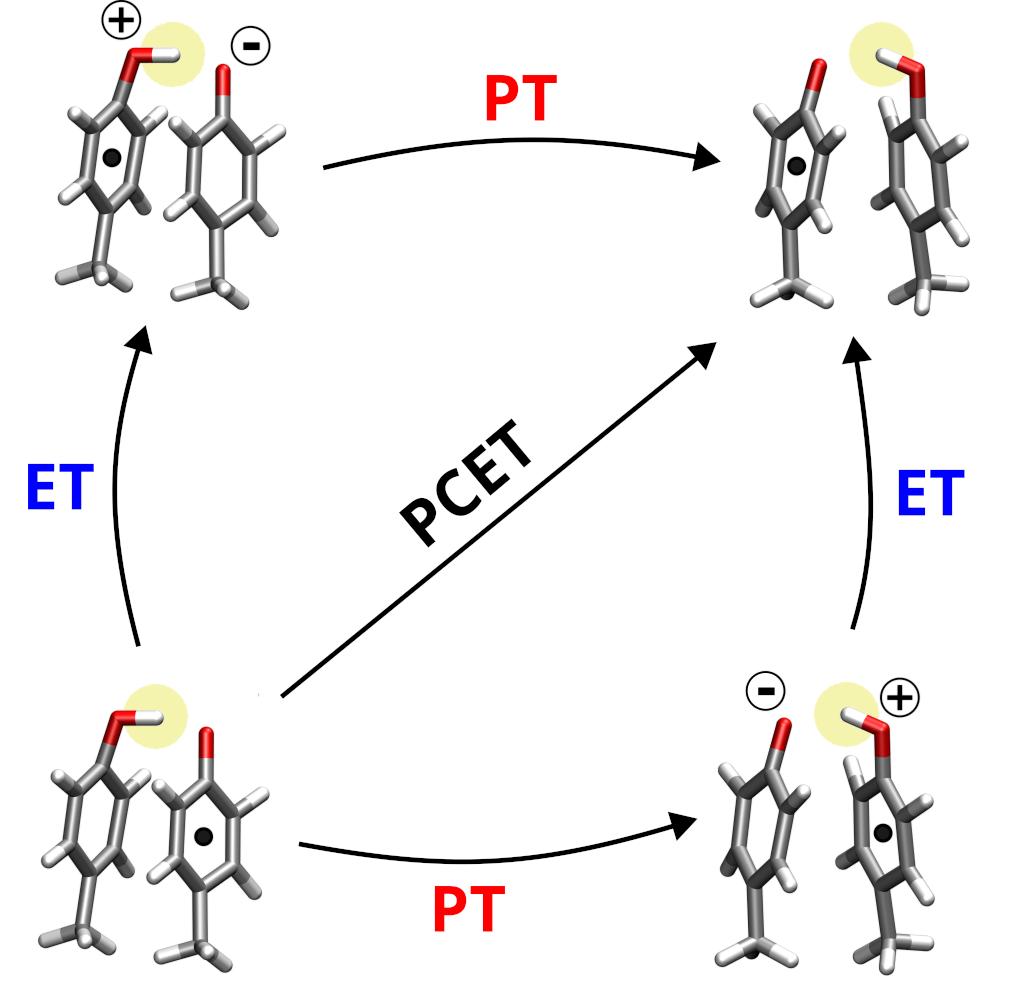
What is PCET?
The transfer of electrons is a key process in energy transduction cascades, which are indispensable components of metabolism in all living organisms. These electrons often move across long distances in complex protein environments, through a chains of individual steps, which are commonly accompanied by the transfer of protons. An elementary reaction that frequently occurs in these processes is a proton-coupled electron transfer (PCET) between aromatic molecules such as the sidechains of tyrosine or tryptophan.
Our aims
We analyze in atomic detail these individual steps that include both electron and proton
transfers. We pay particular attention to the molecular environment of the electron and proton carrying moieties, such as the nearby amino acid residues or the solvent. It turns out that the environments in modulate the PCET strongly, and understanding these effects is essential to explain how the enzymatic energy transduction works.
Identifying Key Collective Variables in High-Dimensional MD Simulations
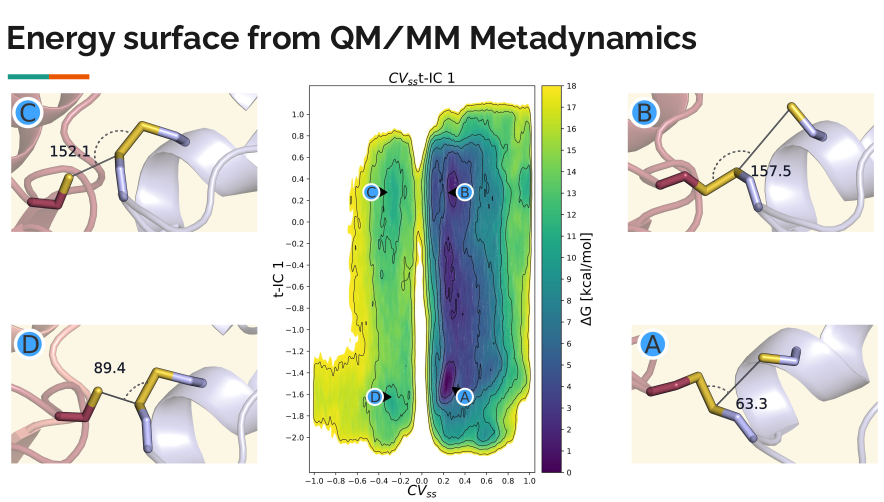
Molecular dynamics (MD) simulations use computational methods to model how atoms and molecules move over time. These simulations generate large amounts of data, capturing complex atomic motions in high-dimensional space. While we can visualize these motions in 3D, the key aspects that define a system's behavior are often hidden within a small set of important reaction coordinates, called collective variables (CVs). For simple chemical reactions, CVs can be chosen intuitively, such as tracking bond formation and breakage. However, for more complex processes, such as protein folding, identifying meaningful CVs is much more challenging and requires advanced methods.
Organic Electronics
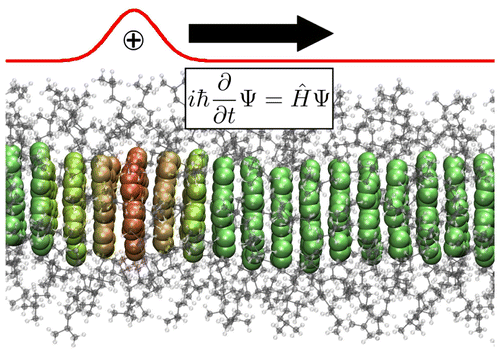
Organic semiconductors (OSCs) have gained prominence as key materials in modern electronics, enabling applications in organic light-emitting diodes (OLEDs), organic photovoltaics (OPVs), and organic field-effect transistors (OFETs). The performance of these devices is governed by charge and exciton transport mechanisms, which are intrinsically influenced by electron-phonon interactions, electronic coupling, and structural disorder. Theoretical and computational methods such as nonadiabatic molecular dynamics (NAMD) and machine-learning-enhanced electronic structure calculations provide insights that extend beyond conventional transport models. Charge transport in OSCs exists in an intermediate regime between band-like and hopping transport. Understanding this crossover requires advanced computational techniques.
Charge transport in OSCs is a complex process influenced by electronic interactions, phonon coupling, and temperature-dependent molecular reorganization. Unlike traditional semiconductors, OSCs exhibit a transport regime that cannot be fully explained by either band-like transport or classical hopping models. Instead, mixed quantum-classical methods such as Fewest-Switches Surface Hopping (FSSH) provide a robust framework for capturing nonadiabatic charge dynamics [1, 2, 3].
A key challenge in modeling charge transport is balancing computational efficiency
with accuracy. To address this, Quantum Mechanics/Molecular Mechanics (QM/MM)
hybrid schemes, combined with efficient fragmentation of the quantum mechanical problem, are employed. In this approach, the charge transport region is treated using semiempirical quantum methods (DFTB), while the surrounding medium is described by molecular mechanics [4]. This strategy enables realistic and scalable simulations of charge propagation in complex organic semiconductor (OSC) materials.
Multiscale computational methods
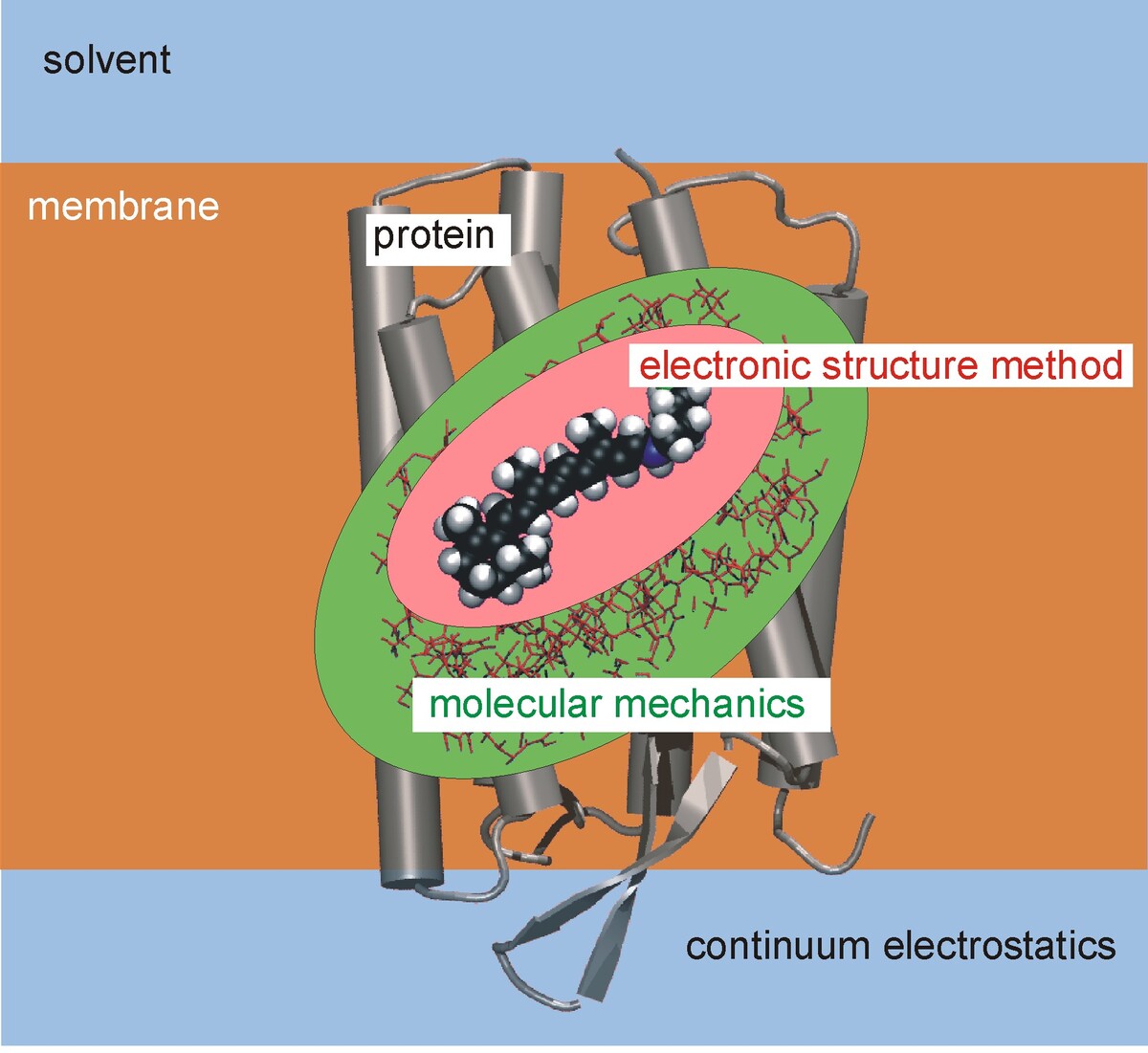
To study chemical reactivity and spectroscopic properties of complex systems like proteins or organic semi-conductors, quantum mechanical (QM) methods are required. Due to the large size of these structures, a simple application of QM methods is impossible due to the high computational cost. Therefore, QM methods are conveniently combined with empirical force field methods (Molecular Mechanics: MM) in the so- called QM/MM methods. In such approaches, the active site of a protein consisting of several tens of atoms is treated with QM, while the remainder of the protein is handled with the computationally cheaper MM methods. This approach can be taken to treat even larger scales by combining the QM/MM methods with continuum approaches. Also, various QM methods with different accuracy can be applied in the active site, building onion-like computation shells of methods.
Besides the development of such method-combinations, a large part of our effort concentrates on the development of fast and accurate approximations to Density Functional Theory (DFT) resulting in the DFTB suite of methods which are about three orders of magnitude faster than DFT, however, keeping a similar accuracy for a broad range of molecular properties.
Topics:
Photo-active proteins
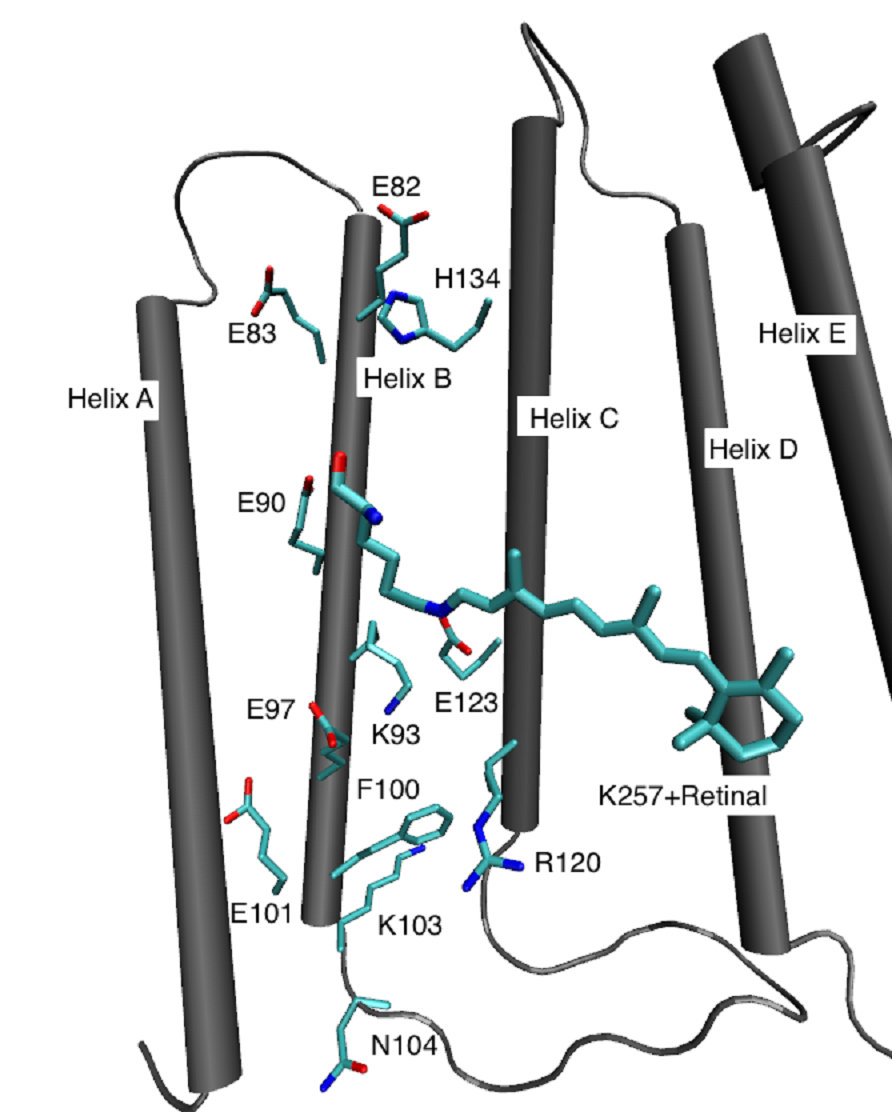
Many proteins make use of the sun light to drive important processes in the cell, like energy generation, or signal conversion. In the last years, we studied intensely the bacterial protein Bacteriorhodopsin (bR), which acts as light driven proton pump. It has been in the focus of several scientific fields for several decades, including various biological, chemical and biophysical approaches, which lead to a detailed understanding of the chemical events and structural changes that accompany the proton pumping process. Simulations, in particular those based on QM/MM methods complemented the picture by computing proton transfer pathways through the protein and energy barriers and relating spectroscopic measurements with the underlying structure.The channelrhodopsins (ChRs) are currently the most regarded members of the rhodopsin proteins and may be considered the most intriguing photo-active proteins discovered in the last decade. They are light-induced ion channels and have drawn significant attention from neuroscientists. They can be readily expressed in a variety of targets, e.g. mammalian neurons, to generate action potentials with light. The ChRs are the foundation of the new method of optogenetics. Despite of their advanced application, surprisingly little is known about their detailed structure and functional mechanism.
Electron Transfer in proteins and organic materials
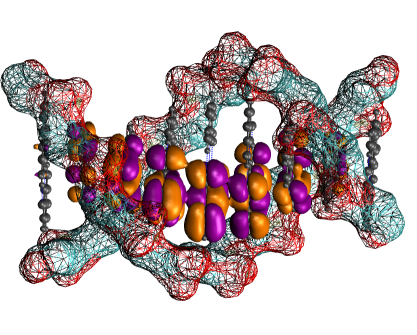
Modeling of electron transfer (ET) events in proteins, DNA and organic materials is a particular challenge for computational chemistry due to the extensive size of the involved QM region on the one hand and to the prolonged time scales on the other. An interesting sub-class of ET processes is those which appear to be relatively fast – on the sub-nanosecond range – and for which the standard approximative schemes like Marcus' theory may no longer be valid. For such processes, we have developed a novel multi-scale methodology which intertwines efficient QM calculations with MM methods and allows for extended sampling of the processes. The electron (or electron hole) is propagated according to the Schrödinger equation within a semi-classical approach while the movement of atoms is described by classical Newton’s equations of motion.
The recent applications are concerned with several topical ET systems. In double-stranded DNA, hole transfer occurs over long distances of up to 100 ns, which has consequences for radiatively induced damage and repair processes. The mechanism of this transfer is controversial to-date, and we are performing simulations designed to resolve this issue. Another application is the hole transfer in the DNA photolyase protein, which takes place in several consecutive steps. It represents a paradigmatic system that violates the assumptions of the classical theory of ET, while our simulations describe the transfer well. A new class of systems that we are starting to work is organic semi-conductors. This project is in an early stage of development, which was made necessary by the fundamental differences between the relevant materials and the biomolecular complexes studied so far.
IV. Interaction of peptides with membranes
Membranes are crucial parts of every cell and a majority of biochemical reactions in metabolism, signalling and transport involves membranes and membrane proteins. Obtaining structural data for anisotropic and complex membrane systems is difficult and computer simulations can help tremendously here.
We study the physico-chemical properties of lipid bilayer biomembrane models using high-efficiency particle dynamics simulations that allow even very slow processes like phase transitions to be described. Both coarse-grained models for large scale processes and atomistic force field molecular dynamics simulations are employed. In collaboration with the group of Prof. Anne Ulrich we draw the connection between simulation results and NMR data to study the mechanism of antimicrobial, membrane penetrating peptides.