
Structure and Function of the Channelrhodopsins
Channelrhodopsins are light-gated cation channels that are part of the control mechanism for the movement of microalgae in response to light (phototaxis, photophobic behavior). Because of their easy expressibility in various host cells and their ability to generate photo currents in a very controlled fashion, they are widely used as tools for the analysis of neural activity in tissues and living organisms (optogenetics).
We want to understand how the channelrhodopsins work and complement experimental efforts to generate modified channelrhodopsins for 1) optimized expression levels, 2) modified channel kinetics, 3) channelrhodopsins with different absorption maxima (color tuning), and 4) modified cation selectivity. Although the application of the channelrhodopsins is considerably advanced, the following fundamental questions are still unanswered:
- Where is the location of the ion channel?
- How is the mechanism for channel opening and closing?
- How is the isomerization of the chromophore (all-trans retinal) connected to the channel mechanism?
- What specific mutations can modify channel kinetics, ion selectivity or absorption maximum?
We want to address these questions by combining computational methods from different fields, such as homology modeling, classical molecular dynamics simulations, QM/MM simulations with fast semi-empirical quantum-mechanical methods, and ab initio quantum-mechanical calculations.
How is the Absorption Maximum of the Chromophore Affected by the Protein Environment? – Color Tuning in the Channelrhodopsins
In order to enhance the optogenetic toolbox, channelrhodopsin variants with shifted absorption maxima are desirable, especially those, which absorb at lower wavelengths (red-shifted). It is well-known from other retinylidene proteins, that the absorption maximum of the retinal can be tuned over a wide range by different protein enviroments.
We examined differences in the binding pocket of the channelrhodopsins with respect to bacteriorhodopsin, because experience gained from other microbial rhodopsins, such as bacteriorhodopsin indicate, that the largest impact on the absorption maximum comes from changes close to the chromophore. We built models of the channelrhodopsins' binding pocket by starting from bacteriorhodopsin and mutating 27 amino acids in the binding pocket. By calculating the excitation energy of the mutants, we evaluated the impact of each mutation on the shift of the absorptiom maximum. By means of simple homology modeling, we found, that the rest of the protein, apart from the binding pocket, has only a minor influence on the absorption maximum.
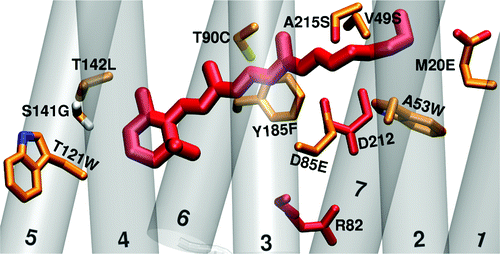
Residues in the binding pocket, that contribute most to the color tuning between the channelrhodopsins and bacteriorhodopsin. Residues marked in red have a large effect on the color tuning (because they are charged) but are conserved between bacteriorhodopsin and the channelrhodopsins.
Welke K. et al.; J. Phys. Chem. B, 2011, 115, 15119
How Does a Channelrhodopsin Look Like? – Structural Model of Channelrhodopsin-2
Channelrhodopsins have seven transmembrane helices. Similarities can be found to other microbial rhodopsins, but are mainly restricted to the last five helices. Helices 1 and 2 are very distinct, so that it is generally believed, that the unique function of the channelrhodopsins are somehow connected to these two helices.
We found a periodicity of charged and polar residues, that are seven sites apart from each other. Based on this periodicity, we built 3D models of Channelrhodopsin-2, which agrees well with experimental data, especially the crystal structure for the channelrhodopsin-1/-2 hybrid. We found large water densities along helix 2 and a hydrophilic core, which is assumed to be a cation-binding site. Additionally, we found remarkable structural motifs, intrahelical hydrogen bonds, that possibly influence the channeling kinetics, as expected from other proteins.
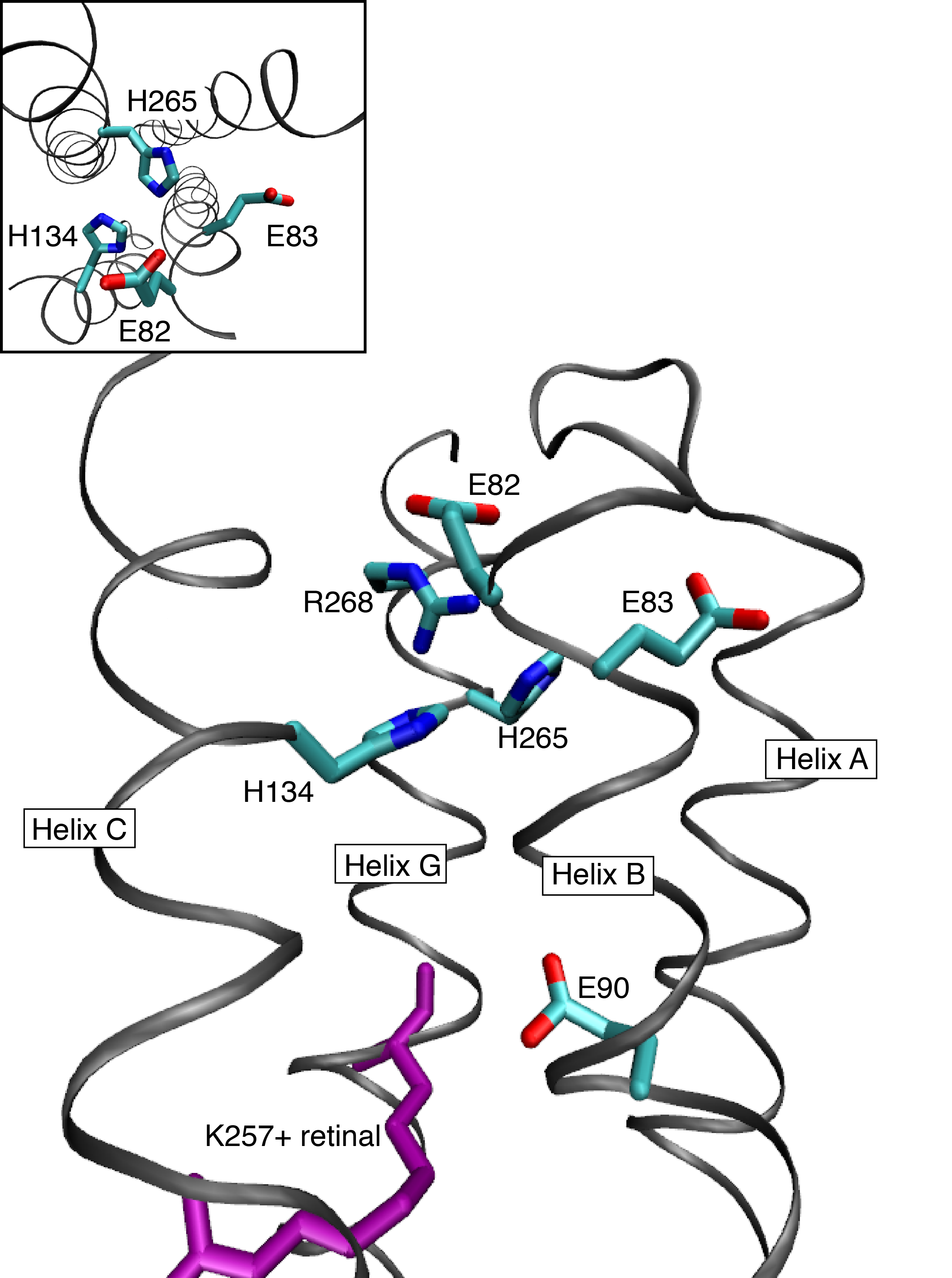
The hydrophilic core found in the cytoplasmic side of channelrhodopsin-2
Watanabe H. C et al.; J. Biol. Chem.; 2012; 287, 7456
How Does The Structure Affect Kinetics? – A Refined Model for Channelrhodopsin-2
We made use of the x-ray structure of the hybrid (first five helices from channelrhodopsin-1, last two helices from channelrhodopsin-2) to generate a refined model for channelrhodopsin-2, which agrees in many aspects with the x-ray structure and our previous homology model.
Several hydrogen bonds we found previously are confirmed to be of functional relevance, since they can lead to helix bending. In this work, we focused on a possible mechanism, how structural changes in the ChR2 active site after photoisomerization of retinal can be transduced to other sites in the protein, finally leading to channel opening. We suggest that the hydrogen-bonding interaction of Thr-127 is crucial for the photocycle. It switches from the side chain of the counterion, Glu-123, to its backbone oxygen, if the counterion is protonated. This change is further transduced to Cys-128 and Asp-156, the so-called DC gate, for which changes have been confirmed with FTIR spectrosopy.
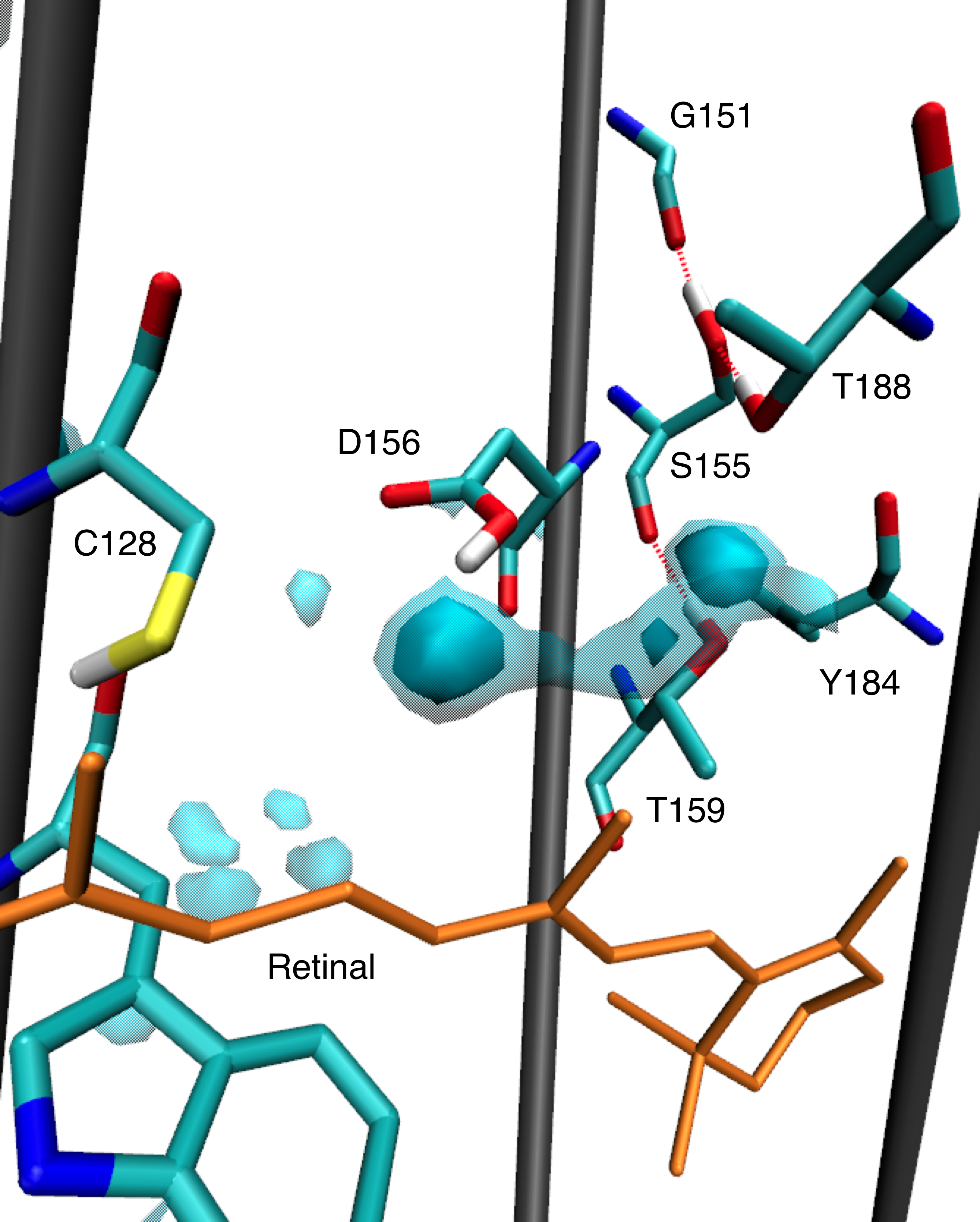
Water distributions found around the DC gate and populated intrahelical hydrogen bon
Watanabe H. C et al.; J. Mol. Biol. 2013; 425; 1795
A Refined Model for the DC Gate in Channelrhodopsin-2 Confirmed by Vibrational Spectroscopic Simulations
The DC gate seems to play a key role in the channel function. Therefore, we dedicated a separate study to provide a better understanding of its structure and spectroscopic properties. Based on observations in FTIR experiments and homology to other microbial rhodopsins, it was concluded, that Asp-156 and Cys-128 are connected via a direct hydrogen bond. However, our simulations predict a water molecule between these residues, not resolved in the x-ray structure and not present in other microbial rhodopsins. We used our model of channelrhodopsin-2 to compute IR spectra using a QM/MM approach. The simulations show, that our proposed model complies with the FTIR data and provides a better explanation of the observations than a direct hydrogen bond.
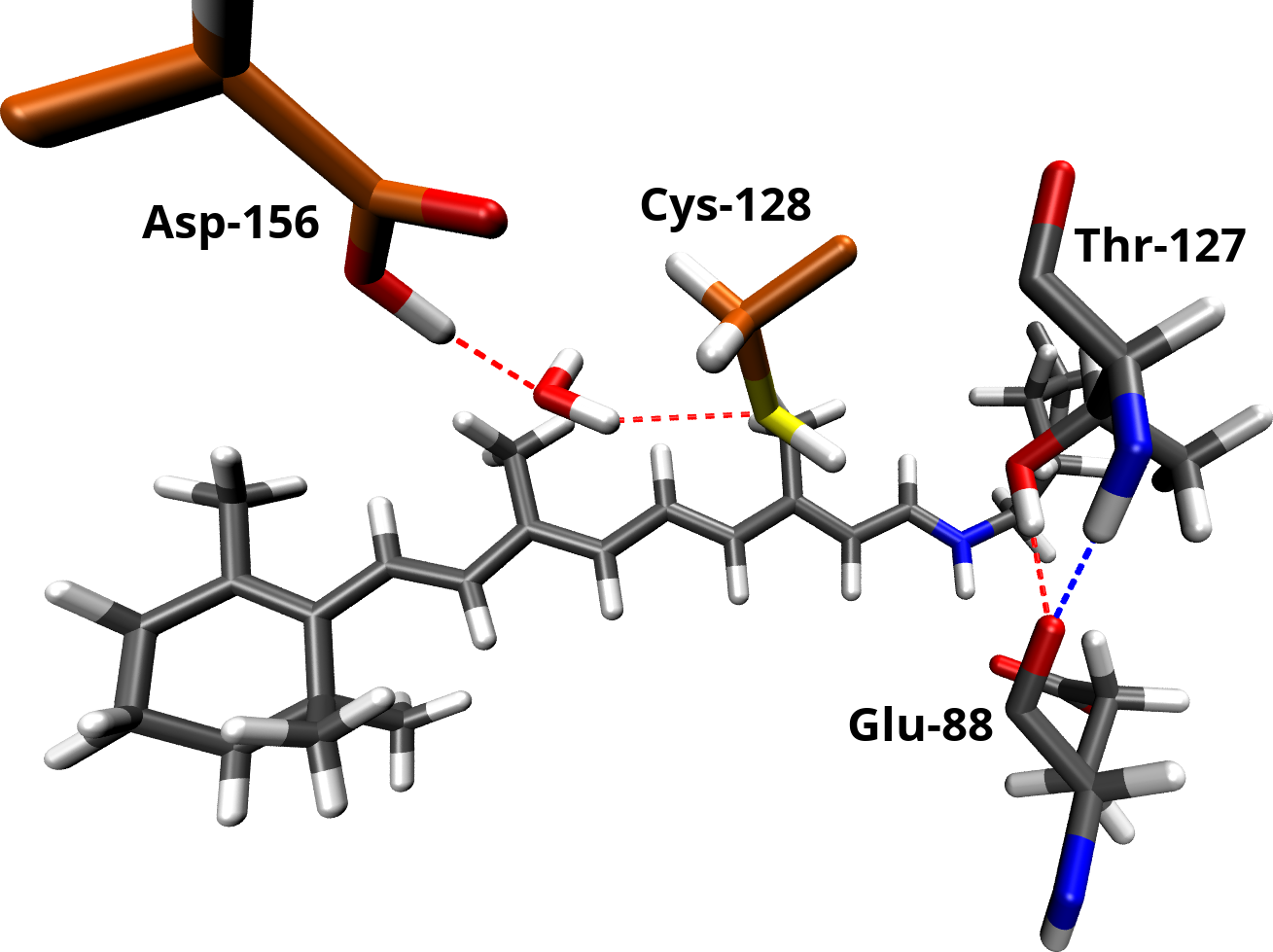
The DC gate in channelrhodopsin-2 consists of Cys-128, Asp-156 and a bridging water molecule. It is connected to the Schiff base region of the retinal vial Thr-127